In response to the significant impact the COVID-19 pandemic is having on European regulatory activity, the European Commission, the European Medicines Agency and the Heads of Medicines Agencies network (EC, EMA and HMA, respectively) have approved a number of measures to help the management of marketing authorisations for human medicinal products considered crucial during the pandemic period.
The objective of these measures agreed at European level is to promote regulatory flexibility, facilitate, simplify and accelerate the administrative procedures, as far as possible, in order to respond more efficiently to emerging needs during this period.
As a result, the EC recently published questions and answers on regulatory expectations for medicinal products for human use during the COVID-19 pandemic:
This Q & A document which provides guidance to marketing authorisation holders (MAH) includes the following topics:
renewal applications
sunset clause
an exceptional change management process (ECMP) for crucial medicines for use in COVID-19 patients
circumstances under which the validity GMP certificates and authorisations to manufacture/import can be extended
circumstances under which the validity GDP certificates and wholesale authorisations can be extended
adaptions to the work of a Qualified Person (QP)
the possibility of adapting quality requirements for medicines intended to be used for the treatment of COVID-19 patients
the impact on reporting into EudraVigilance of Individual Case Safety Reports (ICSRs)
flexibility in the labelling and packaging requirements to facilitate the movement of medicinal products within the EU
Further to the European Commission’s Q&A document, the CMDh has agreed additional questions and answers that provide practical information on how to specifically address and apply the provisions determined by the European Commission for MR/DC procedures:
The CMDh document addresses issues such as the impact of COVID-19 on assessment timelines, how to use the ECMP procedure (which is only applicable for products that are crucial for the treatment of COVID-19 patients) and QP declarations based on a desktop audits. It also includes a useful annex that details Member States’ email addresses and links to relevant published guidance on MS websites.
Both documents will be updated and supplemented with additional information, as appropriate during the pandemic.
Everyone at Ivowen is working tirelessly to keep our clients applications on track. We are liaising with the National Competent Authorities all the time to ensure we avoid delays and get the best results possible in these unprecedented times.
If you need any assistance in this regard please don’t hesitate to contact us.
Written by Claire Brown.
https://eureg.ie/wp-content/uploads/2020/05/Corona-virus-image-11-05-20-1.jpg405720ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2020-05-11 16:24:572023-11-06 11:15:46European Procedural Guidance during COVID-19 Pandemic
Firstly, in this very unusual time for us all I want to let you know that our dedicated team are all healthy, safe and working from home, business as usual, to ensure that we continue to provide you with the high quality and efficient service you have come to expect from Ivowen.
Have you ever wished you could get the opinion of an Assessor before you go to the National Competent Authorities with your queries?
Well, I am delighted to introduce you to to our newest team member Claire Brown.
Claire has come directly to Ivowen from the Health Products Regulatory Authority (HPRA) and brings with her a wealth of experience as a pharmaceutical assessor of human medicinal products. Claire has been part of the team here in Ivowen since 2019 and has more than 12 years experience working in the HPRA. She started there as a Scientific Officer working mostly on veterinary medicinal products and was promoted to Pharmaceutical Assessor after 3 years.
Claire completed her undergraduate work in Chemistry and her postgraduate work in Neuropharmacology and Industrial Pharmaceutical Science.
Claire adds her extensive experience to our knowledgeable team so that we can enhance the services we provide to you and continue to give you an ‘Assessor’s eye’ opinion on your applications before you submit them.
I invite you to visit our updated Meet the Team page to see all the talented people who enable us to guide you through all aspects of regulatory strategy, dossier preparation, MA submissions through to national phase, post-approval variations, product development, quality overviews, medical devices and much much more.
If you could use some assistance to navigate the complex world of regulatory, please feel free to contact us for further information.
Written by Alice D’Alton.
https://eureg.ie/wp-content/uploads/2014/09/pharmacovigilance2.jpg3471080ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2020-03-31 19:02:032023-11-06 11:08:10How to get an Assessor’s opinion before you contact the Competent Authorities
As an MAH, do any of yourauthorised human medicinal products contain chemically synthesised active substances? If so, read the following to identify what your responsibilities are with regard to reporting results of risk analysis to the appropriate Competent Authorities by 26 March 2020 deadline.
Further information for companies in relation to centrally and non-centrally authorised medicines is available from the EMA website and CMDh website.
Background:
In June 2018, authorities in the EU became aware of the presence of a nitrosamine, N-nitrosodimethylamine (NDMA), in valsartan from one manufacturer of active pharmaceutical ingredients (APIs). Subsequently another nitrosamine, N-nitrosodiethylamine (NDEA), was detected and other sartans from more API manufacturers were later implicated.
NDMA and NDEA are classified as probable human carcinogens (substances that could cause cancer) and their presence in sartans was, at the time, unexpected.
An Article 31 review of sartans at risk of containing nitrosamine impurities (i.e. sartans with a tetrazole ring) concluded that manufacturers must review and make necessary changes to their manufacturing processes to minimise nitrosamine impurities as much as possible. In addition, strict limits were set for nitrosamines in these products. The Article 31 review of sartans identified a number of root causes of nitrosamine formation and contamination.
A review was performed which indicated there is a potential for nitrosamines to be present in APIs for other medicines (i.e. non-sartans APIs), depending on the API and the finished product manufacturing processes.
While Nitrosamines are not expected to be formed during the manufacture of the vast majority of APIs outside the class of sartans with a tetrazole ring, it is now known that these impurities can form during production under certain conditions and when certain solvents, reagents and other raw materials are used. In addition, impurities can be carried over during the manufacturing process when using already-contaminated equipment or reagents.
Responsibilities of MAH:
Despite the low risk of nitrosamines being present, Marketing Authorisation Holders (MAHs) are asked to take precautionary measures to mitigate the risk of nitrosamine formation or presence during the manufacture of ALL authorised human medicinal products containing chemically synthesised APIs including generics and over-the counter (OTC) products. However, in view of the large number of authorised products, MAHs should use a risk-based approach and prioritize their evaluations and confirmatory testing.
The EMA’s human medicines committee (CHMP) requested in September 2019 that MAHs for human medicines containing chemically synthesised active substances, review their medicines for the possible presence of nitrosamines and test all products at risk.
It is the responsibility of MAHs to work closely with the manufacturers of APIs and finished products to perform risk evaluation and report this to the Competent Authorities where the products are authorised within 6 months of the guideline on “Information on nitrosamines for marketing authorisation holders”. This guideline was published on 26 September 2019, thus MAHs should supply complete risk evaluations to the respective Competent Authorities by 26 March 2020.
In summary MAHs must perform the following steps:
Step 1: Risk Evaluation
MAH’s should perform risk analysis of their medicinal products containing chemically synthesised API. The MAHs should prioritise products in order to establish the sequence in which their products are to be evaluated. The factors to be taken into account are outlined in the dedicated Q&A document on the EMA & CMDh websites. For products of high priority the risk evaluation should be done immediately. The risk evaluation of all products should be concluded at the latest with 6 months of the publication of Information on Nitrosamines for Marketing Authorisation Holders.
If a risk of presence of nitrosamine is identified as a result of the evaluation, the MAH should proceed to step 2 below.
After the individual risk evaluation is finished send the appropriate template for the outcomes of “no risk identified” or “risk identified” including the required email headings and details. The guidance given by the relevant national competent authorities’ website also has to be regarded, refer to Annex 1 in CMDh practical guidance for MAH of nationally authorised products (including MRP/DCP in relation to Art. 5 (3) Referral for Nitrosamines.
If the presence of nitrosamine is identified as a result of the risk evaluation, confirmatory testing should be carried out using appropriately validated and sensitive methods. Products identified as high priority should be tested as soon as possible.
Confirmatory testing of all medicinal products identified to be at risk of presence of nitrosamines and submission of required changes in the Marketing Authorisations should be concluded within 3 years (by 26/02/2022) of the publication of Information on Nitrosamines for Marketing Authorisation Holders or at an earlier time if otherwise justified.
MAHs should inform the relevant Competent Authorities immediately if tests confirm the presence of nitrosamines impurity irrespective of the amount detected.
MAHs should apply for a variation in a timely manner to introduce any required changes, such as amendment to the manufacturing process or changes to product specifications.
Refer to Templates on the EMA and CMDh websites to report results to the relevant Competent Authorities. It is also advised to view the relevant Competent Authorities websites to view national requirements on submitting this information.
If you need any clarification or support to help implement the responsibilities of a MAH with regard to Nitrosamines reporting contact us and we will gladly assist you in a timely manner.
Written by
https://eureg.ie/wp-content/uploads/2020/02/time-for-action-clock-recd-28-02-20-1.jpg480852ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2020-02-28 16:36:302023-11-06 11:15:31Nitrosamines and responsibilities of MAHs – Deadline is approaching
Like me, you probably get survey requests into your Inbox every day.
Like me, you probably ignore most of them.
Don’t ignore this one!
The European Commission is doing a survey on national regulatory fees for human medicines and it is available to everyone here.
Have your say, and encourage the EC and NCAs to move to an invoicing model, rather than an upfront payment with all the associated admin fees and forms.
https://eureg.ie/wp-content/uploads/2020/02/Survey.jpg8531280ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2020-02-19 11:56:422023-11-06 11:15:42Finally a survey we can all get behind! Regulatory Fees for Human Medicines
The United Kingdom has formally left the EU as of 31 January 2020 and has become what is referred to as a third country.
On 1 February 2020 a transition period started which is due to end on 31 December 2020.
During the transition period, EU pharmaceutical law will continue to be applicable to the UK, meaning that pharmaceutical companies can continue to carry out activities in the UK until the end of 2020.
Companies have until 31 December 2020 to make the necessary changes to ensure that their authorised medicines comply with EU law and can remain on the EU market.
The UK will remain on CESP for the duration of transition period (after that, if no further extension to the transition period is proposed, it will be necessary to use the MHRA portal for submissions https://pclportal.mhra.gov.uk/)
Marketing authorisation holders and applicants can still be established in the UK in 2020
Qualified Persons for Pharmacovigilance (QPPVs) and pharmacovigilance system master files (PSMFs) can still be based in the UK until the end of 2020.
Manufacturing sites, Quality control testing and Batch release sites can also still be based in the UK until the end of 2020
orphan designation holders can still be located in the UK until 31 December 2020
minor use/minor species (MUMS)/limited markets classification holders can still be located in the UK until 31 December 2020
The withdrawal agreement foresees that following its departure from the EU on 31 January 2020, the UK will no longer participate in EU institutions and their decision-making. For the CMDh this means that as of 1 February 2020, no one who represents the UK, or is appointed or nominated by the UK can systemically participate in the CMDh meetings.
During the transition period, the UK will not be able to act as RMS in MRP/DCP, but the UK can participate in MRP/DCP as CMS.
Ivowen are here to assist you with all your Brexit related needs and dossier amendments.
For more information on Ivowen’s services and how we can help you, contact us.
Written by Alice D’Alton
https://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svg00ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2020-02-10 11:40:492023-11-06 11:08:32BREXIT – EVERYTHING stays the same for 2020
Ivowen attended the Medicines for Europe conference in January (Regulatory and Pharmacovigilance), the annual EuDRAcon conference in May, exhibited at TOPRA in October and joined our clients from around the world at CPhI in November.
We all saw Brexit come and go, Twice !! We wait to see what lies in store for the next deadline in January 2020.
The FMD came into effect across Europe in February in most member states.
Bulgaria joined CESP, eCTD became mandatory for all human procedures, lots of new guidance was published (to keep us all on our toes) and Nitrosamines in medicinal products moved to the top of everyone’s agenda.
With the festive season now upon us and 2020 on the horizon, Ivowen are setting our sights on the year ahead.
We will be attending the Medicines for Europe conference in January 2020 (Regulatory and Pharmacovigilance) and we encourage you to contact us before mid-January with any specific questions you might like us to ‘ask the regulators’. This is a great opportunity to ask those difficult questions that you just could not get a straight answer to in 2019, on the ever present grey areas of Regulatory procedures.
To help you to plan ahead here are some helpful updates, in brief, as full articles will be posted in 2020:
Falsified Medicines Directive – Where we are now:
Implemented on 9th Feb 2019 in all MS except Greece, Italy and Belgium
The European Commission has produced a video to explain more about the safety features.
The HPRA have extended the use and learn period, initially to Sep 2019 and extended it again to end on a phased basis starting from 31st January 2020.
The MHRA is also taking a pragmatic, flexible approach to how they enforce the new legal requirements.
Nitrosamines
Step 1: MAHs should conduct a risk evaluation to identify products at risk of N-nitrosamine formation or (cross-)contamination and report the outcome by 26 March 2020 at the latest.
Ivowen are here to assist you in 2020 and will continue to provide the top quality service you have come to expect from us.
For more information on Ivowen’s services and how we can help you, contact us.
Written by Alice D’Alton.
https://eureg.ie/wp-content/uploads/2019/12/Hot_Air_Baloon.jpg356570ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2019-12-06 09:52:522023-11-06 11:08:332019 – What a year
In an ever changing and at times a fast paced environment the world of pharmaceuticals demands cohesion and understanding across the board, to allow growth and success in any business either directly or indirectly involved with it. With regulations and guidelines becoming more stringent and extensive along with constant revisions and updates of the same it can be hard to keep up. It is important that you and your colleagues have an understanding of these guidelines that relate to the work that you do to ensure safety of patients and company compliance.
In certain instances guidelines and recommendations are hundreds of pages long and differ between countries, this places huge time pressure on individuals and businesses to keep up to date and often a brief read over them leaves people feeling a little confused and sometimes without even a basic understanding as to what actually needs to be done.
IVOWEN are here to help you gain a better understanding of, and, confidence in, current guidelines and technical updates. Ivowen has always offered training in many areas of pharmaceuticals but are now looking at providing these training sessions online to help our clients to gain a higher level of expertise through easier access.
We would be very grateful if you could complete the online survey we have created to help us assess how the online training sector can benefit our clients.
It takes less than 3 minutes to complete and would help us immensely:
“No company can afford not to move forward. It may be at the top of the heap today but at the bottom of the heap tomorrow, if it doesn’t.”
— James Cash Penney, founder, JC Penney
Written by Emily Fletcher
https://eureg.ie/wp-content/uploads/2019/11/Help-Support-Advise-Guidance-21-11-19.jpg248372ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2019-11-21 14:01:232023-11-06 11:08:37Are you interested in online training??
This year’s TOPRA Annual Symposium was held in Dublin in October. The Annual Symposium is an essential meeting for regulatory professionals to gain both an understanding of current and evolving regulatory requirements, as well as insights into future plans for regulations in the Human medicines, Veterinary medicines and Medical Device sectors.
Three members of our Team attended the Symposium this year, Majella Ryan, Alice D’Alton and Nanda Naik. This year was particularly satisfying for Ivowen as we had an exhibition stand at the three day event.
As well as exhibiting, Ivowen attended the sessions including ‘Life after Brexit’. This session was a live discussion of predictions and concerns about the possible outcome of ongoing negotiations with the EU.
Now that an extension has been granted, which will delay the UK exit until January 2020, we are left wondering if we will finally know the UK position when Ivowen attend the Medicines for Europe regulatory conference being held in Amsterdam on 29th – 31st January.
In the meantime, we await instruction from the regulators on whether UK will still be available on the CESP portal later this week. Ivowen is registered on the new MHRA submissions portal so either way we have you covered.
Ivowen will keep you up to date in the coming days and weeks
Please contact us, Ivowen are dedicated to keeping you up to date with the latest regulatory updates and innovations. We remain at your disposal to assist in all of your regulatory endeavours today and into the future.
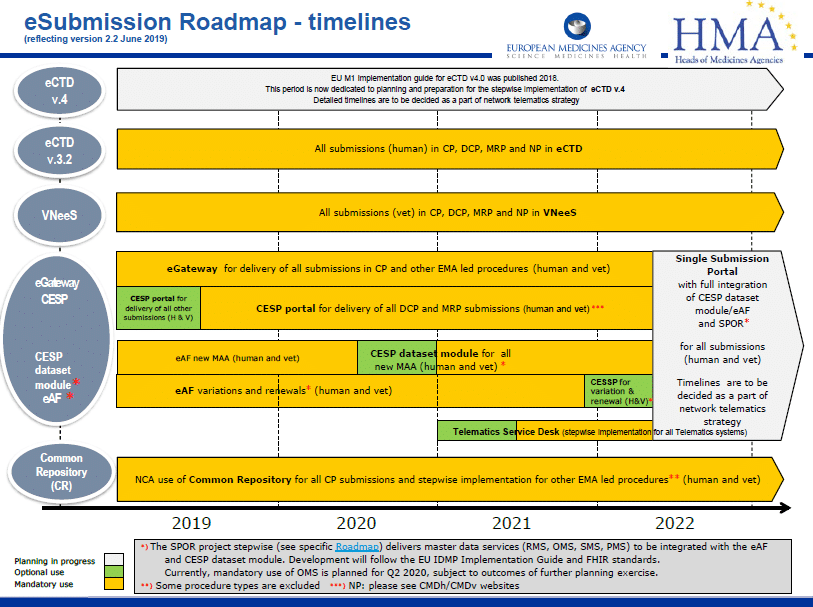
The European Union updated its eSubmission Roadmap in June 2019 to reflect changes in timelines.
What is it?
The eSubmission roadmap is a high level, strategic plan for business and technology changes within the EU. Its function is to align the plans and implementation timelines of target groups and stakeholders, including the EMA, National Competent Authorities (NCAs) and the pharmaceutical industry.
Full details on the eSubmission Roadmap are available on the EMA’s eSubmission website here.
What is happening?
Centralised Procedure(s) (CP)
It is mandatory since January 2010 that all submissions in the CP are made in the eCTD format
National Procedure(s) (MRP & DCP and national only procedures)
It is mandatory since July 2015 and January 2017 that all submissions for new MAAs using the DCP and MRP respectively are made in the eCTD format. For purely national procedures, eCTD has been mandatory for new MAA submissions since July 2018.
For all new submissions in MRP and DCP, the mandatory use of eCTD has been in force since January 2018, and for all new national submissions, since January 2019.
What does this mean?
This means that ALL submissions in the EU for human medicinal products now have to be made in eCTD format. Are you ready? If not, contact us to find out how we can help switch all your current MAs to eCTD and manage all your lifecycle needs. Ivowen has been eCTD compliant since 2009, and therefore has a wealth of experience to become your key partner in this vital step of your registration process.
If you need any clarification or support to manage the changeover to eCTD, Ivowen will gladly work with you to ensure a seamless and efficient transition to eCTD. Contact us for more information or to make an enquiry.
The HPRA are always striving to improve their processes and ways of working. The following updates in the Human Medicines Department should help us all in our dealings with each other.
New electronic workflow system
The human medicines department within the HPRA have transitioned to using a new (internal) electronic workflow system. Due to this the following changes are worth noting for MA (marketing authorisation) applications and the issuing of licences and summary of product characteristics (SmPCs) by the HPRA:
Product Specific Details (PSD)
The product specific information (which included the product composition and the manufacturers’ details) will no longer form part of the product licence document that is issued by the HPRA. Previously the product licence document consisted of the licence cover page, PSD and Summary of Product Characteristics (SmPC). In future, the product licence document will consist of the licence cover page and the SmPC only. The information previously detailed in the PSD will be logged on the HPRA database and remain a registered part of the product marketing authorisation.
Summary of Product Characteristics (SmPC)
Updated SmPCs and Package Leaflets will publish on the website 24 hours after case closure.
(For details relating to the font/format of SmPC documents, further details are found in HPRA newsletter number 62).
PA numbers
PA, or Product Authorisation numbers, are the Irish version of the MA number. Newly allocated PA numbers for new holders will now contain 5 digit prefix.
Case Reference Number (CRN)
Previously CRNs were displayed as seven digits. These will now be alpha numerical for any new cases e.g. CRN00011X. The HPRA will still be able to identify any closed or ongoing cases using the old CRN.
Digital communications
All cases on the new system will be assigned a dedicated e-mail address e.g. [CaseNumber]@case.hpra.ie. This will enable you to send the HPRA case specific communications directly to the case and the allocated team. E-mail correspondence sent to you from the HPRA that is relevant to the case will come from this dedicated e-mail address. The European e-mail boxes will still be used where applicable. You should consult with your IT department to ensure that e-mails of this nature are not blocked in your organisation.
National Scientific Advice Guide
The HPRA have recently updated their National Scientific Advice Guidance (which commenced in 2017) to include additional therapeutic areas for stakeholders. These areas include: anti-infective products, vaccines, disorders of haemostasis and thrombosis, cardiovascular diseases, common allergic conditions, advanced therapies in certain clinical indications and biostatistics. The updated guidance can be found at the following link:
https://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svg00ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2019-08-07 15:22:032023-11-06 10:26:30News from the HPRA