From the implementation of the Windsor Framework on 1 January 2025, the MHRA will regulate medicines across a UK-wide licensing regime.
Most new Marketing Authorisation Applications (MAAs) will be granted as PL licences covering the whole of the UK.
The MA number will have just a PL prefix (as it was before Brexit).
No action is necessary for this conversion for Marketing Authorisation Holders who only hold a PLGB licence for a particular product.
If a MAH currently holds both a PLNI and a PLGB for the same product and wishes to retain UK-wide authorisation, the PLNI will need to be cancelled in order for the existing PLGB to be converted to a PL.
Where an MAH only holds a PLNI and subsequently seeks authorisation for the whole of the UK, the PLNI will need to be cancelled prior to the granting of the PL.
MAHs involved in EU procedures, with NI included as a CMS, will need to notify the RMS to withdraw NI as a CMS from the procedure. This must occur before the cancellation of the PLNI and the granting of the new PL.
‘UK Only’ is required to be printed on the UK packaging from 01Jan2025, a sticker is allowed to be placed on packaging for a period of 6 months (until 30June2025) but this must be replaced with direct printing on the packaging after this.
And from 1 January 2025, the EU Falsified Medicines Directive (FMD) will no longer apply in Northern Ireland.
From 1 January features included for the purposes of compliance with EU FMD requirements may be removed or covered.
MHRA Overview:
Current position
From 1 January 2025
Action required from MAHs
New MAA for product in scope of the Centrally Authorised Procedure
GB: national applications, reliance/recognition applications,
PL
EC no longer able to authorise for NI.
For products entering the market from 1 January 2025, MAHs may apply for PL licences only. Must comply with labelling and packaging requirements.
New MAA for product in scope of the Centrally Authorised Procedure
NI: EC authorisation.
Supply of GB authorised products may be permitted through routes, e.g., NIMAR, Reg 174, Reg 167.
PL
EC no longer able to authorise for NI.
For products entering the market from 1 January 2025, MAHs may apply for PL licences only. Must comply with labelling and packaging requirements.
New MAA for Non-CAP product authorisations
UK-wide authorisation or option to apply for PLGB or PLNI
PL
Or
PLNI through the MRP/DCP
For products entering the market from 1 January 2025, MAHs may apply for PL licences or PLNI only if using the MRP/DCP. Must comply with labelling and packaging requirements.
Products with existing authorisations granted before 1 January 2025.
UK-wide licence
PL
Must comply with labelling and packaging requirements.
Products with existing authorisations granted before 1 January 2025.
Separate PLGB and PLNI for the same product.
Option A:
PL only
By 30 September 2024 the MAH should request that the MHRA cancel the PLNI, effective on 31 December 2024. PLGB licences will convert to PL by 1 January 2025.
Must comply with labelling and packaging requirements.
Products with existing authorisations granted before 1 January 2025.
Separate PLGB and PLNI for the same product.
Option B:
Retain NI only through the MRP/DCP.
No action required from MAH.
PLGB will be cancelled by the MHRA on 31 December 2024 as cannot hold a PL and PLNI simultaneously for the same product.
Must comply with labelling and packaging requirements.
Products with existing authorisations granted before 1 January 2025.
PLNI through the MRP/DCP, with no PLGB for the same product.
PLNI through the MRP/DCP only.
Where a MAH subsequently applies for a PL, the PLNI will need to be cancelled prior to the granting of the PL. The MAH should inform the RMS of its intention to withdraw NI as a CMS from the MRP/DCP.
Must comply with labelling and packaging requirements.
Products with existing authorisations granted before 1 January 2025.
PLGB only
PL only
No action required from MAH.
PLGBs will be converted to PL by 1 Jan 2025.
Must comply with labelling and packaging requirements.
CAP Bridging Mechanism
Permits supply of GB-licensed product in NI for up to 6 months if no equivalent available.
Not applicable as the MHRA will license novel/CAP medicines in NI through a UK-wide licensing route.
Not applicable.
Existing Stock in Existing Packaging on the market
Continues as currently.
Can continue to be supplied to patients until the product’s date of expiry in the territory for which the product was valid for supply prior to 1 Jan 2025.
Not applicable.
More on Windsor Framework in the weeks to come……
If you need any help to navigate this new min field of UK updates, don’t hesitate to contact us for support.
Written by
Emily Fletcher
Emily Fletcher 1
https://eureg.ie/wp-content/uploads/2024/02/Windsor-Framework-recd-13-02-24.jpg640960ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2024-02-13 13:24:342024-02-13 13:25:45UK updates to your Medicine – Windsor Framework and Regulation (EU) 2023/1182
So, let’s start with the Legal Basis which forms the first step of your MA application (MAA for short). You need to know the legal basis before you can finish compiling your application.
The legal requirements and necessary procedures for submitting an initial application for an EU marketing authorisation (MAA) are set out in Directive 2001/83/EC, as amended, and in Regulation (EEC) No. 726/2004 (the latter for CP applications).
The legal requirements for a UK MAA are set out in the Human Medicines Regulations.
The relevant articles and regulations from this legislation are provided below followed by a brief description of each legal basis:
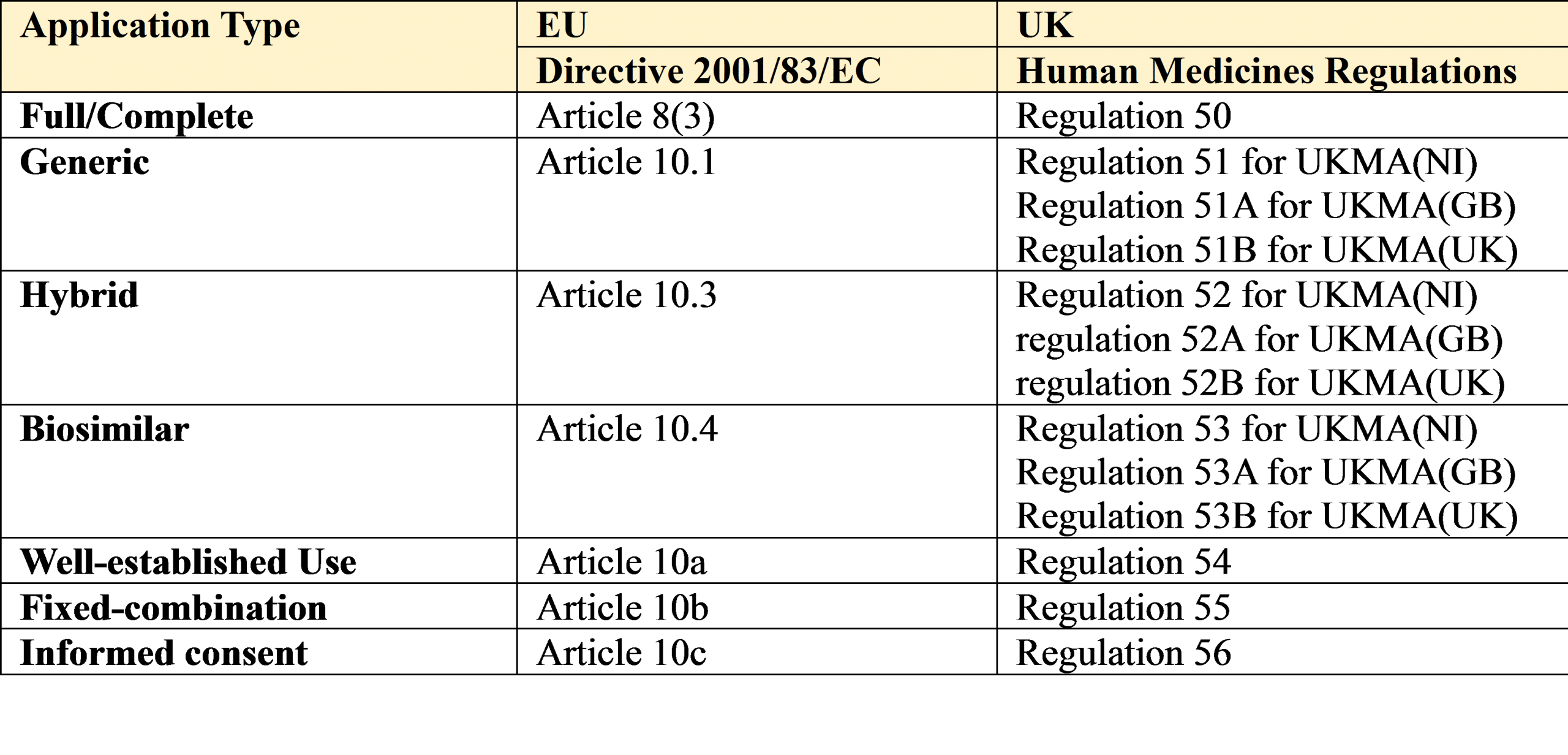
1. Full/Complete
An application for a marketing authorisation (MAA) must be accompanied by the particulars set out in Article 8(3) of Directive 2001/83/EC, as amended, and as well as all the usual documentation will also require the results of
• Physico-chemical, biological or microbiological tests,
• Pharmacological and toxicological tests,
• Full Clinical trials in human subjects
This Legal basis is most often used for brand new medicines (Drug Product) or active substances (Drug Substance) which have never been authorised in EU or UK before. It results in 10 years or 8+2+1 years of data exclusivity. No generic version can be marked until this exclusivity expires.
2. Generic
This is used when the medicinal product (Drug Product) for which an MA is sought is considered essentially similar to and interchangeable with a medicinal product that has already been authorised via Article 8(3) in the EU or UK.
Essentially similar means it has the same active substance, the same amount of active substance (strength), and the same pharmaceutical form.
In this case the Applicant cross refers to an already approved EU/UK reference medicinal product (RMP for short) to use its already approved data on the Pharmaco-toxicological and Clinical aspects of the application and so does not need to repeat these trials. Instead a bioequivalence study in human subjects is required. This data will show that the product is safe and works the same way inside the human body as the RMP does.
3. Hybrid
These are applications which rely on a mixture of data – i.e. a cross reference to the RMP for some aspects of the clinical and pre-clinical data requirement plus provision of additional data in support of any aspect of the proposed drug product which is different to the RMP (i.e. where it is not essentially similar).
This type of application is used when the proposed product has a different/new indication, different/new dosage form, different/new strength, etc. that is not currently approved for the RMP.
4. Biosimilar
These are generic products which are derived from a biological source rather than a chemically synthesised molecule.
It is often difficult to treat biosimilar generic products in the same way as we treat ‘regular’ generics as it is very tricky to prove essential similarity in the conventional ways – therefore provision of some preclinical and clinical data may be necessary to prove safety and efficacy of the biosimilar product.
The Guideline on similar biological medicinal products should be consulted when considering using a non-EEA authorised comparator (i.e. a non- EEA authorised version of the RMP) in the development of a biosimilar.
5. Well Established Use/Bibliographical
It is possible to replace results of pharmacological and toxicological tests or clinical trials with detailed references to published scientific literature if it can be shown that the drug has a ‘well established use’ with recognised efficacy and known safety.
The minimum criterion for using this legal basis of application is that the drug substance concerned has to have been marketed (i.e. in use) in the EU for at least 10 years. It is commonly used when no RMP exists to refer to or to perform a bioequivalence study against.
This type of application, although allowed for under the legislation, is not always accepted by member states (and they have been known to request clinical data, during assessment) but with the use of real-world data and evidence on the increase this type of application may become more common. Only Time will tell.
6. Fixed Combination
In the case of new drug products containing known active ingredients (drug substances), not before used in combination for therapeutic purposes, the results of pharmacological and toxicological tests as well as clinical trials in human subjects, relating to that combination, must be provided.
This legal basis covers entirely new products that are a mixture of two or more drug substances (sometimes called monocomponents), which have already been authorised and marketed in the EU in their own right. Because this is theoretically a full, stand-alone application (not unlike Article 8(3)), the product enjoys the full 10 or 8+2+1 years data of exclusivity.
Please note that the concept of the global marketing authorisation applies here so once a combination is approved any new application(s) for the ‘same’ product cannot be considered new combinations anymore and so an article 10b would not be acceptable in this situation.
7. Informed consent/Duplicate
The Marketing Authorisation Holder (MAH for short) of a complete/standalone dossier (i.e. the originator/innovator/RMP) can consent in writing to allow a new applicant/MAH to refer to the contents of their approved MA (on file with Competent Authority) for the purpose of obtaining a new MA.
The reference dossier must be a complete dossier (Article 8(3)) and consent must be obtained for all three modules containing the pharmaceutical (Module 3), preclinical (Module 4) and clinical data (Module 5).
The Applicant must choose the correct legal basis in advance of submission and selecting the wrong one will delay your application.
If you need any clarification or support contact us and we will gladly assist you.
If you are starting out in Regulatory Affairs and you now have a long list of questions after reading this article, we can help. We provide detailed training for all levels of Regulatory experience. You or your manager can contact us for details and our training menu.
Written by
Fiona Downey 1
https://eureg.ie/wp-content/uploads/2023/11/Legal-basis.jpg10002000ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2023-11-23 20:50:292023-11-23 20:54:32Back to Basics – Regulatory 101 – Legal basis of your MA application in the EU and UK
The new veterinary regulation (NVR), Regulation 2019/6 applied to all EU Member States from 28 January 2022. The new legislation represents a significant change in how veterinary medicinal products are authorised, monitored and controlled in the EU.
The Regulation was developed in order to implement a fit-for-purpose veterinary legislation which would no longer be based on the equivalent human medicines authorisation system.
The new Regulation 2019/6 is broken down into the following chapters:
I. Subject matter, scope and definitions
II. Marketing authorisations
III. Procedures for marketing authorisations
IV. Post marketing authorisation measures
V. Homeopathic veterinary medicinal products
VI. Manufacturing, import and export
VII. Supply and use
VIII. Inspections and controls
IX. Restriction sand penalties
X. Regulatory network
XII. Common and procedural provisions
XII. Transitional and final provisions
Here is a summary of some of the noteworthy regulatory changes that have been introduced in chapters II, III & IV of the new veterinary regulation:
Chapter II – Marketing Authorisations:
An MA for a veterinary medicinal product shall be valid for an unlimited period of time. Hence, there is no longer a requirement for a renewal procedure or the sunset clause.
Chapter III – Procedures for marketing authorisations
Decentralised Procedure:
Scope and timelines remain unchanged
Responsibilities of RMS, CMS and applicant have changed at some steps of the procedure. For example,
– CMSs will also provide comments directly to the applicant at Day 100 and Day 145 instead of to the RMS only (therefore, comments are no longer anonymised).
– at Day 100-105 and Day 145-150, the applicant will now compile and circulate the LoQs.
– at Day 210, RMS will now be required to circulate a Final Assessment Report (FAR).
Scope remains the same. However, a minimum of six months is required between the decision granting the national MA and submission of an application for a MRP.
90 day procedure length remains unchanged but there are changes to some of the time-points in the procedure.
Responsibilities of RMS, CMS and applicant have changed at some steps of the procedure, similar to those outlined above for the DCP.
Centralised Procedure:
Scope of the mandatory use of the procedure has been widened. Refer to Article 42 (point no. 4) for details.
National Procedure:
No significant changes.
Subsequent Recognition Procedure (SRP):
Previously known as the “Repeat Use Procedure” is now officially recognised under Article 53 of the NVR.
Timelines and other requirements have been changed.
Due to the changes caused by the new regulation, CMDv have published updated guidance for DCP, MRP and SRP procedures:
Chapter IV: Post marketing authorisation measures (Variations)
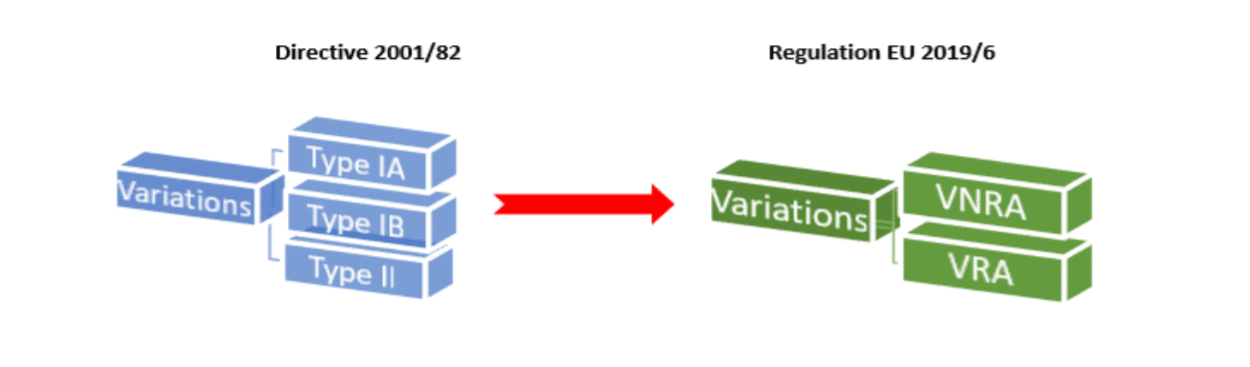
In terms of variations to marketing authorisations, one of the main changes arising from Regulation EU 2019/6 is that instead of the previous categories of Type IA, IB and II variations there will now only be two categories of variations:
Variations Not Requiring Assessment = VNRA
Variations Requiring Assessment = VRA
VNRAs consist of all the previous type IA and some Type IB variation categories.
VRAs will consist of most of the previous Type IB and all of the Type II variation categories.
Commission Regulation 1234/2008 will now no longer apply to veterinary medicinal products due to the introduction of new veterinary regulation (2019/6).
Variations Not Requiring Assessment (VNRA)
The Implementing Regulation (EU) 2021/17*, includes a list of all variations not requiring assessment along with any associated conditions and documentation requirements and is published in the EU Official Journal here.
The variations are classified as follows:
Administrative changes
Changes to the quality part of the dossier
Changes to the safety, efficacy and pharmacovigilance part of the dossier
Changes to the vaccine antigen master file (VAMF) part of the dossier
VNRAs will be processed as follows:
The MAH will:
– Record the change in the Product Union Database (UPD) within 30 days of implementation including required documents (no application form is necessary).
– Documents submitted directly to UPD. No CESP submission. Documents include those listed in the Implementing Act as well as SPC, package leaflet, labels.
The relevant CA/RMS will
– Approve/reject the variation
– Inform MAH & CMS by recording decision in database and by e-mail
The format and categorisation is similar to the previous regulation which applied (Commission Regulation 1234/2008 ), however there are many differences.
The variations are divided into chapters as follows:
E. Administrative changes
F. Quality changes
G. Safety, Efficacy and Pharmacovigilance changes
H. VAMF or, PTMF changes
I. Changes of active substance(s), strength, pharmaceutical form, route of administration or food producing target species
Z-categories have also been included to address unlisted variations and VNRA, if requirements laid down in the Implementing Regulation are not met.
The timetable for VRAs is also outlined in the new guidance as follows:
a standard timetable, denoted by ‘S’ which will be 60 days
a reduced timetable, denoted by ‘R’ which will be 30 days
an extended timetable, denoted by ‘E’ which will be 90 days
For details on how to submit a VRA, CMDv have written a Best Practice Guide for Variations Requiring Assessment (EMA/CMDv/144277/2021). It has been prepared in order to facilitate the processing of VRAs for MRP/DCP products. The same general principles will apply to purely nationally authorised products.
Recommendation for the classification of variations not already listed
A procedure for requesting a recommendation for the classification of variations not already listed in either the above-mentioned Implementing Regulation or the EMA/CMDv Guidance on variations requiring assessment has also been established. This is similar to the previous CMDv recommendations for classification of unforeseen variations, according to Article 5 of Regulation 1234/2008.
Grouping and worksharing procedures do not apply to VNRA, they only apply to VRA.
As a consequence, no VNRAs can be included in a grouping or worksharing even if they are consequential or related to the VRAs included in the grouping or worksharing procedure.
However, the introduction section of CMDv Best Practice Guide for Variations Requiring Assessment (which also covers grouping), outlines the different approaches to follow when there is a need to co-ordinate changes that are related or consequential but are classified as VNRA and VRA.
The worksharing procedure is outlined in Article 65 of the NVR and it will be compulsory to follow this procedure, when the same change is being applied in different member states. Information related to worksharing is also mentioned in the CMDv BPG for Variations Requiring Assessment. However a specific guide on worksharing has also been written by CMDv: Best Practice Guide for Worksharing (EMA/CMDv/204024/2021).
Union Product Database
Due to the new regulation, the EMA has introduced new IT systems. The main one will be theUnion Product Database (UPD).
It will contain information for all authorised veterinary medicines in the EU (including all nationally authorised products). For MAHs, it will provide self-service access for specific regulatory activities, including the management of variationsthat do not require assessment.
For more information on implementation, training, registration and access of the UPD, refer to the following link here on the EMA website.
The UPD will be linked to the other 3 other databases in the future. These databases are at different stages of development and introduction:
Union Pharmacovigilance Database
On 28 January 2022, the Union Pharmacovigilance Database (EVV) was successfully released. User guidance and the release notes are available here.
Manufacturing and Wholesale Distribution Database
The Manufacturers and Wholesale Distributors database (MWD) was released on 28 January 2022. The system is an enhanced and upgraded version of EudraGMDP, the EU database of manufacturing authorisations and certificates of good manufacturing practice, with changes affecting both the veterinary and the human domains. The MWD Project Group has also adopted requirements for aligning the GDP module with the change made to the system so far. Changes to the module will be delivered in a subsequent release scheduled for Q1 2022. In addition, enhanced search facilities on the GMP module will be delivered in the same release.
Database for the Collection of Data on Sales and Use of Antimicrobials in Animals.
IT development on the Collection of Antimicrobials Sales and Use Data (ASU) project started in January 2022. Information on the progress of this project will be published on the EMA website as this project develops.
Q&A on transitional arrangements
CMDv has prepared a Q&A document in order to assist both MAHs and NCAs in the management of the transition between the requirements of Directive 2001/82/EC and Regulation (EU) 2019/6. This Q&A document will be regularly updated and can be found under the following link.
This document includes an Annex which outlines how individual Member States will handle renewals of marketing authorisations after 28 January 2022.
Should you need any support with Veterinary Procedures feel free to contact us & the Ivowen team will be here to help.
Written by Claire Brown
https://eureg.ie/wp-content/uploads/2022/04/Vet-article-feature-image-05-04-22.jpg6221106ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2022-04-05 12:52:052023-11-06 11:18:32The new veterinary regulation (Regulation 2019/6) and its implications on regulatory submissions for veterinary medicinal products
There are now several routes to obtain a marketing authorisation in the United Kingdom (UK), Great Britain (England, Scotland and Wales) or Northern Ireland. The options available will be determined by the intended market and the type of application. In this article we will discuss the purely National procedure.
To help you to decide what type of license you will require, here is a brief explanation of the new types of MA you can obtain:
MA prefix for UK
Possible MA types
Territory
Leg & Guidance
PL
UK wide
Authorised for use in United Kingdom (Great Britain & Northern Ireland)
EU & MHRA rules apply
PLGB
GB
Authorised for use in Great Britain only (England, Scotland and Wales only)
MHRA rules apply
PLNI
NI
Authorised for use in Northern Ireland only
EU rules apply
One option you can pursue is the National procedure (a 150-day procedure) to obtain a marketing authorisation (MA) in the UK, Great Britain or/in Northern Ireland. The MHRA has introduced this accelerated procedure aimed at expediting the availability of medicines for patients in the UK and proposes to reach its opinion on marketing authorisation applications (MAAs) within 150 days of filing an application (excluding the time taken to provide further information or data required).
The accelerated assessment is available for all high-quality new MAAs for both new, and existing active substances, as well as orphan designations. Interested applicants should contact the MHRA in advance of submitting the application.
For medicines containing new active substances or biosimilar products, the MHRA encourages applicants to provide a summary of the dossier to share their intentions and to verify the new active substance status.
The pre-submission meeting offers the opportunity to discuss the arrangements for the UK Compliance Check (CC) on Paediatric Investigation Plans (PIPs). Additionally, it also offers the opportunity to enhance joint discussion with National Institute for Health and Care Excellence (NICE) Health Technology Assessment (HTA) evaluation process.
The MHRA will operate a ‘fixed submission date’ system to facilitate consultation with the Commission on Human Medicines (CHM) and will publish a set of dates to facilitate planning the submissions to coordinate with appropriate meeting dates of CHM. The submission slots will be linked to the dates of CHM meetings.
The assessment timetable will begin after the validation of the application. The assessment process will run in two phases totalling 150 days like so:
Phase I: completed 80 days after the clock starts. Issues that arose or requiring clarification from the initial assessment will be raised with the applicant and should be addressed within the clock off period of 60 days.
Phase II: commence on receipt of the applicant’s responses. The MHRA will provide a decision on the acceptability of the product by day 150.
If the MHRA refuses to grant the MA-based on advice from CHM, there is an opportunity for the applicant to request a review of the decision.
The conclusion of the assessment will lead to the publication of a UK Public Assessment Report for the product.
Here are some useful links to obtain further information:
If you need any clarification or support to help you to navigate the new post Brexit procedures, please contact us and Ivowen will gladly assist you in a timely manner.
https://eureg.ie/wp-content/uploads/2019/01/Brexit2.jpg413640ivowenadminhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgivowenadmin2021-04-21 13:25:462023-11-06 11:15:36New UK National Procedure – Expedited 150 day procedure
The UK has left the EU and the transition period after Brexit comes to an end this year.
The MHRA have issued new guidance for industry and organisations effective from 01st January 2021. From this date the MHRA will be the UK’s standalone medicines and medical devices regulator.
Areas covered in the new guidance include:
Clinical Trials
From 1 January 2021, for registering clinical trials, existing and established international registers will still be used, such as ISRCTN registry (UK), or ClinicalTrials.gov (USA), to ensure the public is aware of your trial. For trials involving both UK and EU sites a record in the EU Clinical Trials Register will exist (other than adult Phase 1 studies). In the UK, any favourable opinion given by a research ethics committee is subject to the condition that the clinical trial is registered on a publicly accessible database. The time frame for publishing the summary of results is within 6 months of the end of trial for paediatric clinical trials or within one year of the end of trial for non-paediatric clinical trials. You do not need to submit this clinical trial summary report to the MHRA as well; however, you must send a short confirmatory email to CT.Submission@mhra.gov.uk once the result-related information has been uploaded to the public register and provide a link.
Pharmacovigilance
Guidance on qualified person responsible for pharmacovigilance (QPPV) including pharmacovigilance system master files (PSMF) from 1 January 2021
From 1 January 2021, the following legal obligations will apply to holders of UK marketing authorisations (MA). These include those that cover the whole of the UK, or are specific to Northern Ireland or to Great Britain (England, Wales and Scotland):
To operate a pharmacovigilance system for UK authorised products.
To have an appropriately qualified person responsible for pharmacovigilance (QPPV) that resides and operates in the EU or the UK and is responsible for the establishment and maintenance of the pharmacovigilance system for UK authorised products.
To maintain and make available upon request a pharmacovigilance system master file (PSMF) that describes the pharmacovigilance system for UK authorised products. The PSMF must be accessible electronically or physically from the UK at the same site at which reports of suspected adverse reaction may be accessed.
Statutory guidance concerning the QPPV for UK authorised products is described in the Good Pharmacovigilance Practices (GVP) Module I. This guidance will be supplemented by the ‘Exceptions and modifications to the EU guidance on good pharmacovigilance practices that apply to UK marketing authorisation holders’, which will be published in due course.
New guidelines have been outlined for Marketing Authorisations, to include Conditional MAs, registering new packaging information, guidance on the handling of applications for Centrally Authorised Products (CAPs), Article 29 applications, converting parallel distribution notices to UK parallel import licences, handling of ASMFs and CoS from January 2021, reference medicinal products, converting CAPs to UK MAs, guidance on licencing biosimilars, bioequivalence/therapeutic equivalence studies and renewing marketing authorisations.
New Submission Registrations
For planned applications for submission to the UK (for example, a Marketing Authorisation for the UK market), you will need to submit the information through the MHRA national portals.
All current Eudravigilance Gateway users who wish to gain access to the new MHRA Gateway will need to first gain access to MHRA Submissions. The steps for gaining MHRA Gateway access are contained within MHRA Submissions. MHRA Submissions will not be used to send or receive ICSRs.
If you need any clarification or support to help you to navigate the end of transition period please contact us and Ivowen will gladly assist you in a timely manner.
Written by Mary Canning
https://eureg.ie/wp-content/uploads/2020/07/Brexit-clock-image-22-07-20.jpg422750ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2020-11-06 12:15:002023-11-06 11:15:33BREXIT – MHRA post-transition period information
Did you know, that pharmaceutical companies can consult the European Medicines Agency (EMA) to determine whether a medicine they are developing is considered an advanced therapy medicinal product (ATMP)?
In doing so, they are allowed to receive confirmation that a medicine, which is based on genes, cells or tissues, meets the scientific criteria for defining an ATMP. The criteria for ATMPs are set out in Article 17 of Regulation (EC) No 1394/2007.
In order to submit an application for ATMP classification, applicants have to complete a:
pre-submission request form
ATMP-classification request form and briefing information
and return both forms to the EMA.
Applies to your company? Ivowen are here to assist you.
For more information on Ivowen’s services and how we can help you, contact us.
Firstly, in this very unusual time for us all I want to let you know that our dedicated team are all healthy, safe and working from home, business as usual, to ensure that we continue to provide you with the high quality and efficient service you have come to expect from Ivowen.
Have you ever wished you could get the opinion of an Assessor before you go to the National Competent Authorities with your queries?
Well, I am delighted to introduce you to to our newest team member Claire Brown.
Claire has come directly to Ivowen from the Health Products Regulatory Authority (HPRA) and brings with her a wealth of experience as a pharmaceutical assessor of human medicinal products. Claire has been part of the team here in Ivowen since 2019 and has more than 12 years experience working in the HPRA. She started there as a Scientific Officer working mostly on veterinary medicinal products and was promoted to Pharmaceutical Assessor after 3 years.
Claire completed her undergraduate work in Chemistry and her postgraduate work in Neuropharmacology and Industrial Pharmaceutical Science.
Claire adds her extensive experience to our knowledgeable team so that we can enhance the services we provide to you and continue to give you an ‘Assessor’s eye’ opinion on your applications before you submit them.
I invite you to visit our updated Meet the Team page to see all the talented people who enable us to guide you through all aspects of regulatory strategy, dossier preparation, MA submissions through to national phase, post-approval variations, product development, quality overviews, medical devices and much much more.
If you could use some assistance to navigate the complex world of regulatory, please feel free to contact us for further information.
Written by Alice D’Alton.
https://eureg.ie/wp-content/uploads/2014/09/pharmacovigilance2.jpg3471080ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2020-03-31 19:02:032023-11-06 11:08:10How to get an Assessor’s opinion before you contact the Competent Authorities
The United Kingdom has formally left the EU as of 31 January 2020 and has become what is referred to as a third country.
On 1 February 2020 a transition period started which is due to end on 31 December 2020.
During the transition period, EU pharmaceutical law will continue to be applicable to the UK, meaning that pharmaceutical companies can continue to carry out activities in the UK until the end of 2020.
Companies have until 31 December 2020 to make the necessary changes to ensure that their authorised medicines comply with EU law and can remain on the EU market.
The UK will remain on CESP for the duration of transition period (after that, if no further extension to the transition period is proposed, it will be necessary to use the MHRA portal for submissions https://pclportal.mhra.gov.uk/)
Marketing authorisation holders and applicants can still be established in the UK in 2020
Qualified Persons for Pharmacovigilance (QPPVs) and pharmacovigilance system master files (PSMFs) can still be based in the UK until the end of 2020.
Manufacturing sites, Quality control testing and Batch release sites can also still be based in the UK until the end of 2020
orphan designation holders can still be located in the UK until 31 December 2020
minor use/minor species (MUMS)/limited markets classification holders can still be located in the UK until 31 December 2020
The withdrawal agreement foresees that following its departure from the EU on 31 January 2020, the UK will no longer participate in EU institutions and their decision-making. For the CMDh this means that as of 1 February 2020, no one who represents the UK, or is appointed or nominated by the UK can systemically participate in the CMDh meetings.
During the transition period, the UK will not be able to act as RMS in MRP/DCP, but the UK can participate in MRP/DCP as CMS.
Ivowen are here to assist you with all your Brexit related needs and dossier amendments.
For more information on Ivowen’s services and how we can help you, contact us.
Written by Alice D’Alton
https://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svg00ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2020-02-10 11:40:492023-11-06 11:08:32BREXIT – EVERYTHING stays the same for 2020
The HPRA are always striving to improve their processes and ways of working. The following updates in the Human Medicines Department should help us all in our dealings with each other.
New electronic workflow system
The human medicines department within the HPRA have transitioned to using a new (internal) electronic workflow system. Due to this the following changes are worth noting for MA (marketing authorisation) applications and the issuing of licences and summary of product characteristics (SmPCs) by the HPRA:
Product Specific Details (PSD)
The product specific information (which included the product composition and the manufacturers’ details) will no longer form part of the product licence document that is issued by the HPRA. Previously the product licence document consisted of the licence cover page, PSD and Summary of Product Characteristics (SmPC). In future, the product licence document will consist of the licence cover page and the SmPC only. The information previously detailed in the PSD will be logged on the HPRA database and remain a registered part of the product marketing authorisation.
Summary of Product Characteristics (SmPC)
Updated SmPCs and Package Leaflets will publish on the website 24 hours after case closure.
(For details relating to the font/format of SmPC documents, further details are found in HPRA newsletter number 62).
PA numbers
PA, or Product Authorisation numbers, are the Irish version of the MA number. Newly allocated PA numbers for new holders will now contain 5 digit prefix.
Case Reference Number (CRN)
Previously CRNs were displayed as seven digits. These will now be alpha numerical for any new cases e.g. CRN00011X. The HPRA will still be able to identify any closed or ongoing cases using the old CRN.
Digital communications
All cases on the new system will be assigned a dedicated e-mail address e.g. [CaseNumber]@case.hpra.ie. This will enable you to send the HPRA case specific communications directly to the case and the allocated team. E-mail correspondence sent to you from the HPRA that is relevant to the case will come from this dedicated e-mail address. The European e-mail boxes will still be used where applicable. You should consult with your IT department to ensure that e-mails of this nature are not blocked in your organisation.
National Scientific Advice Guide
The HPRA have recently updated their National Scientific Advice Guidance (which commenced in 2017) to include additional therapeutic areas for stakeholders. These areas include: anti-infective products, vaccines, disorders of haemostasis and thrombosis, cardiovascular diseases, common allergic conditions, advanced therapies in certain clinical indications and biostatistics. The updated guidance can be found at the following link:
https://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svg00ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2019-08-07 15:22:032023-11-06 10:26:30News from the HPRA
Brexit extension date is now 31 October 2019, but there’s not time to be complacent.
Are you planning to submit a Marketing Authorisation Application (MAA) for a Non-Prescription Medicine?
If you are preparing an MAA for a Non-Prescription Medicine for a Decentralised (DCP) or Mutual Recognition Procedure (MRP) you will need to prepare a ‘Justification for Non-Prescription Classification’ document in accordance to CMDh best practice guide issued in January 2019 (CMDh/250/2012, Rev 1).
Ideally this document should be submitted as part of the initial MAA. Otherwise, you will be requested to provide it at the validation stage.
This document should be titled, “Justification for Non-Prescription Classification” and should be provided in Module 1.2. It should contain all the supporting data and evidence required to justify classification of the medicinal product as not subject to medical prescription as set out in the Guideline on Changing the Classification for the Supply of a Medicinal Product for Human Use [https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-2/c/switchguide_160106_en.pdf].
If you need any clarification or advise to prepare a justification for Non-Prescription classification report to support a Marketing Authorisation Application, Ivowen will gladly assist you in a timely manner. Please contact us.
Related news in relation to Brexit
The current Brexit extension date is 31 October 2019. The UK remains an EU Member State for the duration of the extension, with all the rights and obligations set out in the treaties and under EU law.
Nevertheless, all pharmaceutical companies in the EU are reminded to continue their preparedness for the UK’s withdrawal.
Based on the European Council decision, the deadline of 29 March 2019 referred to in Brexit related guidance should be understood as referring to 31 October 2019.
The MHRA have updated guidance on Exporting active substance manufactured in the UK in a no deal scenario on 03/06/2019.
In the event of a no deal EU exit, the UK will be recognised as a Third Country for the export of active substances for human use to the EEA.
In the event of a no deal scenario, the UK will continue to accept importation of active substances into the UK without a Written Confirmation from the same list of countries as currently (namely the European Economic Area (EEA) countries, USA, Japan, Brazil, Australia, Israel and Switzerland).
A Written Confirmation will then be required for each shipment of active substances manufactured in the UK that is exported to the EEA.
Please contact us if you need any clarification or support to supply Written Confirmation(s) for the orderly import of Active Substances, Ivowen will gladly assist you in a timely manner.
The MHRA have issued a guidance on the Handling of Active Substance Master Files and Certificates of Suitability in the event of no deal published 18 March 2019.
After Brexit, the UK will no longer participate in ASMF work-sharing procedures with EU Member States or have access to the EU Communication and Tracking System (CTS) assessment report repository. Any reference in the above guideline to the CTS ASMF assessment repository or to EU/ASMF/XXXXX reference numbers will not be applicable to UK national applications after the UK leaves the EU.
Certificates of Suitability (CEPs) are not affected by the UK leaving the EU as they are issued by the European Directorate for the Quality of Medicines and Healthcare (EDQM), which is a Directorate of the Council of Europe and a body that is independent of the EU. On leaving the EU, the UK will remain a member of the Council of Europe and a signatory to the Convention on the Elaboration of a European Pharmacopoeia.
Please contact us if you need any additional information or if you need any clarification or advise on ASMFs or CEPs, Ivowen will gladly assist you in a timely manner.